
Jan. 29, 2007 — If you bend a knee or an elbow, the nerves in your limbs stretch but do not break. A University of Utah study suggests why: A gene produces a springy protein that keeps nerve cells flexible. When the gene was disabled in tiny nematode worms, their nerve cells literally broke.
The discovery may provide a new explanation for spinocerebellar ataxia type 5 (SCA5) – a disease previously tied to a human version of the gene and identified in 11 generations of U.S. President Abraham Lincoln’s family, starting with his paternal grandparents. SCA5 may have afflicted Lincoln himself. The new study suggests how.
“Were Lincoln’s nerves shattered? We don’t know. But our study raises the possibility that they were,” says biology Professor Michael Bastiani, the study’s senior author and a member of the Brain Institute at the University of Utah.
The new study will be published in The Journal of Cell Biology on Jan. 29, two weeks before Lincoln’s birthday on Feb. 12.
The study involved a worm gene named unc-70 that makes a protein named beta spectrin. Humans have four beta spectrin genes, and mutations in one of them was identified previously as the cause of SCA5, a neurodegenerative disease that develops between ages 10 and 68; destroys nerve cells in the part of the brain that controls movements; causes loss of coordination in walking, speaking, writing and swallowing; and puts some patients in wheelchairs.
Other researchers previously suggested the disease occurs because the mutation leaves beta spectrin unable to anchor other proteins to their proper places in nerve cells, so the cells cannot communicate normally. But the worm study suggests the true cause is nerve-cell breakage caused by defective beta spectrin, says study co-author and biology Professor Erik Jorgensen, scientific director of the Brain Institute at the University of Utah and an investigator with the Howard Hughes Medical Institute (HHMI).
Because the mutant worms lack beta spectrin while human SCA5 patients have an altered form of the protein, more research will be needed to settle the debate, he adds.
A Broken Path to Nerve Cell Diseases?
Earlier research showed beta spectrin makes red blood cells flexible so they don’t burst, and that people with a beta spectrin mutation suffer a form of anemia.
The Utah biologists showed in nematode worms – frequently used as genetic stand-ins for humans – that loss of beta spectrin leaves neurons (nerve cells) prone to breakage. They found the microscopic protein acts like a tiny Slinky spring toy within the coating of nerve cells, giving them strength.
When 1-millimeter-long nematode worms were bred with the mutant gene, they lacked beta spectrin, resulting in breakage of their nerve cells’ axons – the long, wire-like extensions that connect nerve cells. The worms suffered increasing paralysis, then died.
“It’s incredible and so very simple that this one protein is what keeps neurons from breaking in your body,” Bastiani says. “The entire functioning of the nervous system depends on these wire-like axons between nerve cells.”
Jorgensen says the mutant gene also may be involved in other neurodegenerative diseases – he preferred not to speculate on which diseases – and in brain damage that results from stroke or sports injuries.
“In the late stages of a variety of neurodegenerative diseases, you get irreversible nerve cell loss, and it’s possible that is due to loss of this springy protein and nerve elasticity,” says study co-author Marc Hammarlund, a University of Utah postdoctoral researcher in biology and HHMI research associate.
That means protecting spectrin in nerve cells potentially could serve as a treatment to slow progression of some neurodegenerative diseases, he adds.
How a Good Gene Gone Bad May Cause ‘Lincoln’s disease’
Last year, University of Minnesota researchers reported they examined 299 living descendants of the Lincoln family and found the mutant beta spectrin gene in 125 of them, and 90 already had symptoms of what family members call “Lincoln’s disease.”
Those researchers reported there was a one-in-four chance the 16th president had spinocerebellar ataxia type 5. They cited a journalist’s 1861 description of the tall, lanky Lincoln’s “shambling, loose, irregular, almost unsteady gait.”
Jorgensen says Lincoln – who was born in 1809 and became president in 1861 – may have had early-stage SCA5 that had not progressed much by the time he was assassinated in 1865 at age 56.
Most of a nerve cell’s length is the thin, wire-like axon. A typical axon’s diameter is about 5 microns in humans, or one-twentieth the thickness of a human hair. Every movement we make – not just flexing a knee or other joint – “puts stress on neurons,” Bastiani says. Yet the thin axons do not break.
Other researchers have suggested nerve cells also must be flexible to survive the process of wiring the nervous system. As a neuron grows, it sends out a “growth cone” which pulls the growing axon behind it. The axon must be able to withstand stretching.
How it does that has been a mystery – until now.
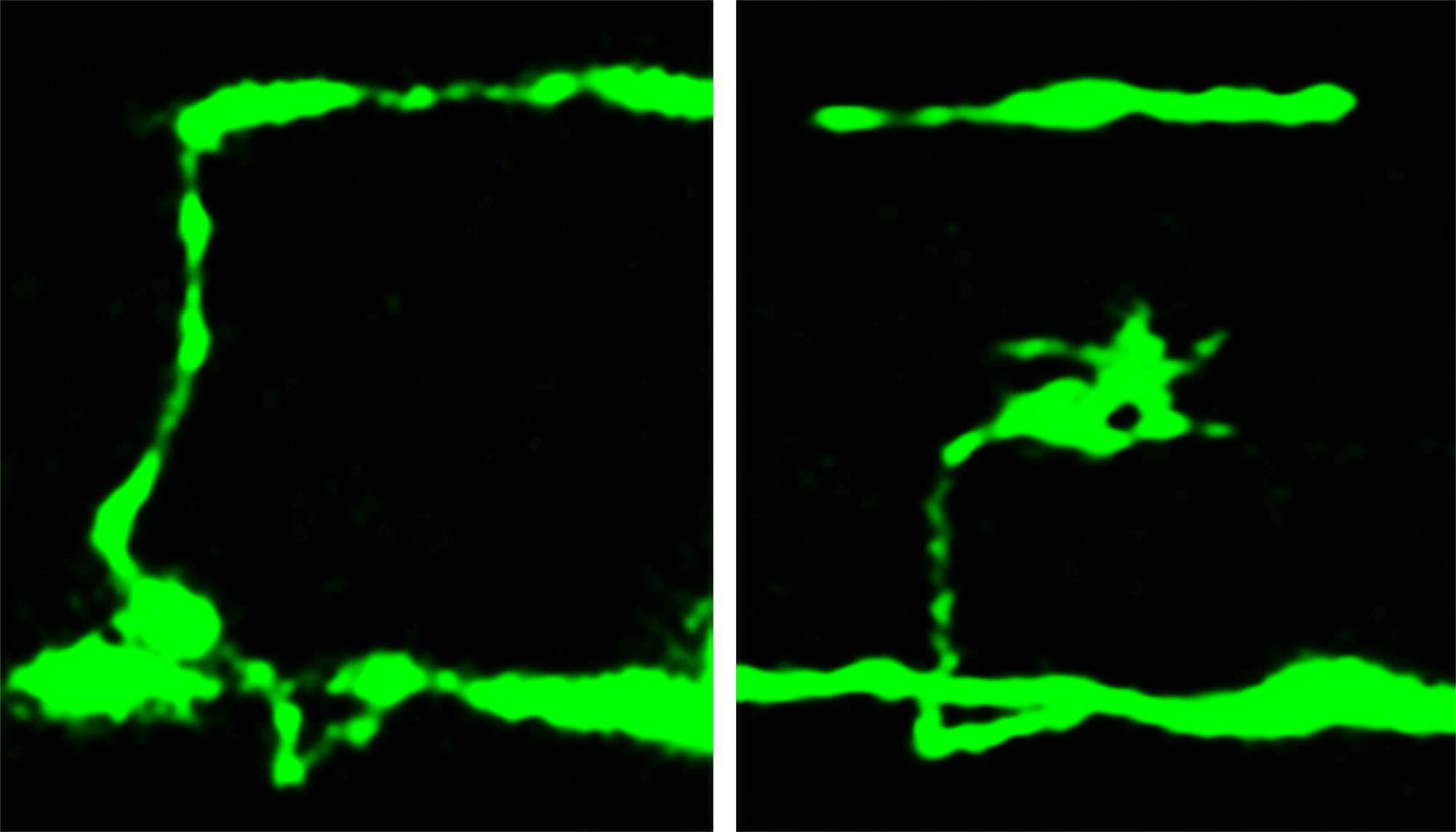
In the new study, the Utah biologists used mutant nematode worms lacking beta spectrin. The study focused on how the protein’s absence affected axons, which carry nerve signals away from the nerve cell “body” and toward the synapse – the junction with another nerve cell.
The researchers used microscopes to photograph nerve-cell axons by inserting a jellyfish gene into the worms to make the worms’ neurons glow fluorescent green.
The biologists found breaks as well as branching or inappropriate new growth cones – signs of nerve-repair attempts – in places where a nerve cell body sent an axon from one side of the worm to the other, making the axon and any damage easy to see.
The neurons were examined at three stages: as embryos, just after the worms hatched and at one day of age. The worms and their nerve cells still were growing during the first two stages.
In worm embryos, only 3 percent of nerve cells were broken or defective, despite the lack of beta spectrin. But in newly hatched mutant worms, 26 percent of nerve cells were broken or defective, and the percentage rose to 60 percent in day-old mutant worms.
Because the nerves did not break in younger worms, beta spectrin is not required for normal nerve growth and development, but is needed for preventing breakage of mature neurons, Hammarlund says.
In another experiment, the biologists paralyzed the worms by disabling two genes needed for muscles to contract. “In paralyzed worms, the axons do not break, even in the absence of spectrin, which says it is movement that causes the nerve cells to break, rather than continued growth of the animal,” Jorgensen says.
That means beta spectrin normally prevents breakage by protecting neurons from the strain of movement, and not by helping add new membrane to a growing nerve cell.
Bastiani calls it amazing that disease-causing nerve-cell damage may come in the form of broken axons – the simplest part of a nerve cell. “Everybody focuses on the synapse or the cell body as to where the action is,” he says. “But if you break the wire or axon, the neuron doesn’t work anymore. It’s sort of the weakest link in the process.”